Nilotinib Capsules is indicated for the treatment of adult patients with newly diagnosed Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase. Additional pediatric use information is approved for Novartis Pharmaceuticals Corporation’s Tasigna® (nilotinib) capsules. However, due to Novartis Pharmaceuticals Corporation’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
Nilotinib Capsules is indicated for the treatment of adult patients with chronic phase and accelerated phase Philadelphia chromosome positive chronic myelogenous leukemia (Ph+ CML) resistant or intolerant to prior therapy that included imatinib. Additional pediatric use information is approved for Novartis Pharmaceuticals Corporation’s Tasigna® (nilotinib) capsules. However, due to Novartis Pharmaceuticals Corporation’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
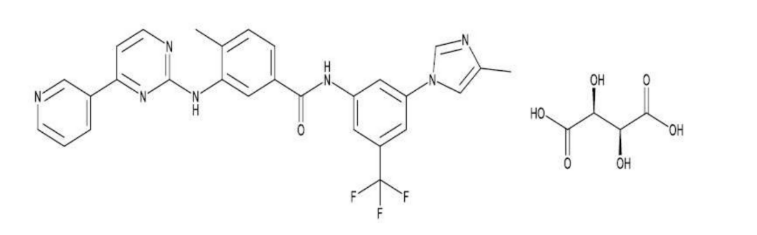
Nilotinib Capsules contains nilotinib d-tartrate, which belongs to a pharmacologic class of drugs known as kinase inhibitors. Nilotinib d-tartrate is a white to yellow powder with the molecular formula and weight, respectively, of C32H28F3N7O7 and 679.61 g/mol (corresponding molecular formula and weight of nilotinib base, anhydrous are C28H22F3N7O and 529 g/mol, respectively). The solubility of nilotinib d-tartrate in aqueous solutions decreases with increasing pH. The pKa1 of nilotinib d-tartrate was determined to be 4.0. The chemical name of nilotinib d-tartrate is 4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3- ((4-(pyridin-3-yl)pyrimidin-2-yl)amino)benzamide D-Tartrate. Its structure is shown below:
Nilotinib Capsules, for oral use, contain 50 mg, 150 mg, or 200 mg nilotinib, anhydrous (equivalent to 64.172 mg, 192.517 mg, and 256.689 mg nilotinib d-tartrate, respectively). The inactive ingredients of Nilotinib Capsules are colloidal silicon dioxide, crospovidone, lactose monohydrate, magnesium aluminometasilicate, magnesium stearate, and polysorbate 80. The inactive ingredients in the empty capsules contain gelatin, iron oxide (black), iron oxide (red), iron oxide (yellow), and titanium dioxide. The black ink contains iron oxide (black), potassium hydroxide, propylene glycol, shellac, and strong ammonia solution.
- 50 mg: light yellow colored blend filled in hard gelatin capsule size 4 cap red opaque linear imprinted with 'Cipla' in black ink and body white opaque linear imprinted with ‘030 50 mg’ in black ink. Each capsule contains the equivalent of 50 mg of nilotinib.
- 150 mg: light yellow colored blend filled in hard gelatin capsule size 1, cap red opaque linear imprinted with 'Cipla' in black ink and body red opaque linear imprinted with ‘031 150 mg’ in black ink. Each capsule contains the equivalent of 150 mg of nilotinib.
- 200 mg: light yellow colored blend filled in hard gelatin capsule size 0, cap white opaque linear imprinted with ‘Cipla’ in black ink and body white opaque linear imprinted with ‘032 200 mg’ in black ink. Each capsule contains the equivalent of 200 mg of nilotinib.
Recommended Dosage
Dosage in Adult Patients with Newly Diagnosed Ph+ CML-CP . The recommended dosage of Nilotinib Capsules is 300 mg orally twice daily. Dosage in Adult Patients with Resistant or Intolerant Ph+ CML-CP and CML-AP The recommended dosage of Nilotinib Capsules is 400 mg orally twice daily.
Additional Administration Instructions
Dose Nilotinib Capsules twice daily at approximately 12-hour intervals on an empty stomach. No food should be
consumed for at least 2 hours before the dose is taken and for at least 1 hour after the dose is taken. Advise patients to
swallow the capsules whole with water and not open the capsules [see Boxed Warning, Clinical Pharmacology (12.3)].
If a dose of Nilotinib Capsules is missed, the patient should take the next scheduled dose at its regular time. The patient
should not take two doses at the same time.
Optional Concomitant Therapy
Nilotinib Capsules may be given in combination with hematopoietic growth factors, such as erythropoietin or G-CSF if
clinically indicated. Nilotinib Capsules may be given with hydroxyurea or anagrelide if clinically indicated.
Additional pediatric use information is approved for Novartis Pharmaceuticals Corporation’s Tasigna®
(nilotinib)
capsules. However, due to Novartis Pharmaceuticals Corporation’s marketing exclusivity rights, this drug product is
not labeled with that pediatric information.
Discontinuation of Treatment After a Sustained Molecular Response (MR4.5) on Nilotinib Capsules
Patient Selection Eligibility for Discontinuation of Treatment
Ph+ CML-CP patients with typical BCR-ABL transcripts, who have been taking Nilotinib Capsules for a minimum of 3
years and have achieved a sustained molecular response (MR4.5, corresponding to = BCR-ABL/ABL ≤ 0.0032% IS),
may be eligible for treatment discontinuation [see Clinical Studies (14.3, 14.4)]. Information on FDA authorized tests
for the detection and quantitation of BCR-ABL transcripts to determine eligibility for treatment discontinuation is
available at http://www.fda.gov/CompanionDiagnostics.
Patients with typical BCR-ABL transcripts (e13a2/b2a2 or e14a2/b3a2), who achieve the sustained MR4.5 criteria, are
eligible for discontinuation of Nilotinib Capsules. Patients must continue to be monitored for possible loss of molecular
remission after treatment discontinuation. Use the same FDA-authorized test to consistently monitor molecular response
levels while on and off treatment.
- been treated with Nilotinib Capsules for at least 3 years
- maintained a molecular response of at least MR4.0 (corresponding to = BCR-ABL/ABL ≤ 0.01% IS) for one year prior to discontinuation of therapy
- achieved an MR4.5 for the last assessment taken immediately prior to discontinuation of therapy
- been confirmed to express the typical BCR-ABL transcripts (e13a2/b2a2 or e14a2/b3a2)
- no history of accelerated phase or blast crisis
- no history of prior attempts of treatment-free remission discontinuation that resulted in relapse.
Consider discontinuation in patients with Ph+ CML-CP that are resistant or intolerant to imatinib who have achieved a sustained molecular response (MR4.5) on Nilotinib Capsules who have:
- been treated with Nilotinib Capsules for a minimum of 3 years
- been treated with imatinib only prior to treatment with Nilotinib Capsules
- achieved a molecular response of MR4.5 (corresponding to = BCR-ABL/ABL ≤ 0.0032% IS)
- sustained an MR4.5 for a minimum of one year immediately prior to discontinuation of therapy
- been confirmed to express the typical BCR-ABL transcripts (e13a2/b2a2 or e14a2/b3a2)
- no history of accelerated phase or blast crisis
- no history of prior attempts of treatment-free remission discontinuation that resulted in relapse.
Monitor BCR-ABL transcript levels and complete blood count (CBC) with differential in patients who have discontinued Nilotinib Capsules therapy monthly for one year, then every 6 weeks for the second year, and every 12 weeks thereafter [see Warnings and Precautions (5.16)].
Upon the loss of MR4.0 (corresponding to = BCR-ABL/ABL ≤ 0.01% IS) during the treatment-free phase, monitor BCR-ABL transcript levels every 2 weeks until BCR-ABL levels remain lower than major molecular response [(MMR), corresponding to MR3.0 or = BCR-ABL/ABL ≤ 0.1% IS] for 4 consecutive measurements. The patient can then proceed to the original monitoring schedule.
Reinitiation of Treatment in Patients Who Lose Molecular Response After Discontinuation of Therapy With Nilotinib Capsules
- Newly diagnosed patients who lose MMR must reinitiate treatment within 4 weeks at the dose level prior to discontinuation of therapy [see Warnings and Precautions (5.16)]. Patients who reinitiate Nilotinib Capsules therapy should have their BCR-ABL transcript levels monitored monthly until major molecular response is re-established and every 12 weeks thereafter.
- Patients resistant or intolerant to prior treatment that included imatinib with confirmed loss of MR4.0 (2 consecutive measures separated by at least 4 weeks showing loss of MR4.0) or loss of MMR must reinitiate treatment within 4 weeks at the dose level prior to discontinuation of therapy [see Warnings and Precautions (5.16)]. Patients who reinitiate Nilotinib Capsules therapy should have their BCR-ABL transcript levels monitored monthly until previous major molecular response or MR4.0 is re-established and every 12 weeks thereafter.
Dosage Modification for QT Interval Prolongation
See Table 2 for dose adjustments for QT interval prolongation [see Warnings and Precautions (5.2), Clinical Pharmacology (12.2)].
Table 2: Dosage Adjustments for Adult Patients With QT Prolongation
| Degree of QTc prolongation | Dosage adjustment |
|---|---|
| ECGs with a QTc greater than 480 msec |
|
Abbreviation: ECG, electrocardiogram. |
Additional pediatric use information is approved for Novartis Pharmaceuticals Corporation’s Tasigna® (nilotinib) capsules. However, due to Novartis Pharmaceuticals Corporation’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
Dosage Modifications for Myelosuppression
Withhold or reduce Nilotinib Capsules dosage for hematological toxicities (neutropenia, thrombocytopenia) that are not related to underlying leukemia (Table 3) [see Warnings and Precautions (5.1)].
Note:
For additional references to the above information, please reference the Prescribing Information
Overdose with nilotinib has been reported, where an unspecified number of nilotinib were ingested in combination with alcohol and other drugs. Events included neutropenia, vomiting, and drowsiness. In the event of overdose, observe the patient and provide appropriate supportive treatment.
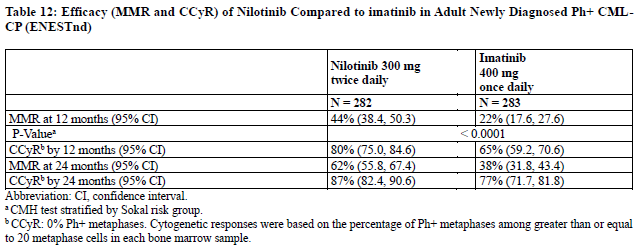
Adult Newly Diagnosed Ph+ CML-CP The ENESTnd (Evaluating Nilotinib Efficacy and Safety in clinical Trials-Newly Diagnosed patients) study (NCT00471497) was an open-label, multicenter, randomized trial conducted to determine the efficacy of nilotinib versus imatinib tablets in adult patients with cytogenetically confirmed newly diagnosed Ph+ CML-CP. Patients were within 6 months of diagnosis and were previously untreated for CML-CP, except for hydroxyurea and/or anagrelide. Efficacy was based on a total of 846 patients: 283 patients in the imatinib 400 mg once daily group, 282 patients in the nilotinib 300 mg twice daily group, 281 patients in the nilotinib 400 mg twice daily group (an unapproved dosage regimen for this indication). Median age was 46 years in the imatinib group and 47 years in both nilotinib groups, with 12%, 13%, and 10% of patients greater than or equal to 65 years of age in imatinib 400 mg once daily, nilotinib 300 mg twice daily and nilotinib 400 mg twice daily treatment groups, respectively. There were slightly more male than female patients in all groups (56%, 56%, and 62% in imatinib 400 mg once daily, nilotinib 300 mg twice daily and nilotinib 400 mg twice daily treatment groups, respectively). More than 60% of all patients were White, and 25% were Asian.
The primary data analysis was performed when all 846 patients completed 12 months of treatment (or discontinued earlier). Subsequent analyses were done when patients completed 24, 36, 48, and 60 months of treatment (or discontinued earlier). The median time on treatment was approximately 61 months in all three treatment groups. The primary efficacy endpoint was major molecular response (MMR) at 12 months after the start of study medication. MMR was defined as less than or equal to 0.1% BCR-ABL/ABL % by international scale measured by RQ-PCR, which corresponds to a greater than or equal to 3 log reduction of BCR-ABL transcript from standardized baseline. Efficacy endpoints are summarized in Table 12.
Two patients in the nilotinib arm progressed to either accelerated phase or blast crisis (both within the first 6 months of treatment) while 12 patients on the imatinib arm progressed to either accelerated phase or blast crisis (7 patients within first 6 months, 2 patients within 6 to 12 months, 2 patients within 12 to 18 months and 1 patient within 18 to 24 months).
Case Studies
14.2 Adult Patients With Resistant or Intolerant Ph+ CML-CP and CML-AP
Study CAMN107A2101 (referred to as Study A2101) (NCT00109707) was a single-arm, open-label, multicenter study
conducted to evaluate the efficacy and safety of nilotinib (400 mg twice daily) in patients with imatinib-resistant or -
intolerant CML with separate cohorts for chronic and accelerated phase disease. The definition of imatinib resistance
included failure to achieve a complete hematologic response (by 3 months), cytogenetic response (by 6 months) or major
cytogenetic response (by 12 months) or progression of disease after a previous cytogenetic or hematologic response.
Imatinib intolerance was defined as discontinuation of treatment due to toxicity and lack of a major cytogenetic response
at time of study entry. At the time of data cutoff, 321 patients with CML-CP and 137 patients with CML-AP with a
minimum follow-up of 24 months were enrolled. In this study, about 50% of CML-CP and CML-AP patients were males,
over 90% (CML-CP) and 80% (CML-AP) were White, and approximately 30% were age 65 years or older.
Overall, 73% of patients were imatinib resistant while 27% were imatinib intolerant. The median time of prior imatinib
treatment was approximately 32 (CML-CP) and 28 (CML-AP) months. Prior therapy included hydroxyurea in 85% of
patients, interferon in 56% and stem cell or bone marrow transplant in 8%. The median highest prior imatinib dose was
600 mg per day for patients with CML-CP and CML-AP, and the highest prior imatinib dose was greater than or equal
to 600 mg/day in 74% of all patients with 40% of patients receiving imatinib doses greater than or equal to 800 mg/day.
Median duration of nilotinib treatment was 18.4 months in patients with CML-CP and 8.7 months in patients with CMLAP.
The efficacy endpoint in CML-CP was unconfirmed major cytogenetic response (MCyR) which included complete and
partial cytogenetic responses.
The efficacy endpoint in CML-AP was confirmed hematologic response (HR), defined as either a complete hematologic
response (CHR) or no evidence of leukemia (NEL). The rates of response for CML-CP and CMLAP patients are reported
in Table 13.
Median durations of response had not been reached at the time of data analysis.
13: Efficacy of Nilotinib in Adult Resistant or Intolerant Ph+ CML-CP and CML-AP (Study A2101)
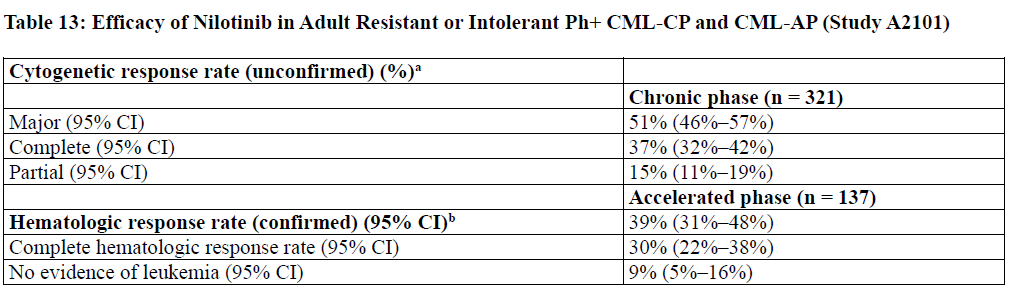
a Cytogenetic response criteria: Complete (0% Ph+ metaphases) or partial (1% to 35%). Cytogenetic responses were based on the
percentage of Ph-positive metaphases among greater than or equal to 20 metaphase cells in each bone marrow sample.
b Hematologic response = CHR + NEL (all responses confirmed after 4 weeks).
CHR (CML-CP): WBC less than 10 x 109/L, platelets less than 450,000/mm3, no blasts or promyelocytes in peripheral blood, less
than 5% myelocytes + metamyelocytes in bone marrow, less than 20% basophils in peripheral blood, and no extramedullary
involvement. CHR (CML-AP): neutrophils greater than or equal to 1.5 x 109/L, platelets greater than or equal to 100 x 109/L, no
myeloblasts in peripheral blood, myeloblasts less than 5% in bone marrow, and no extramedullary involvement.
NEL: same criteria as for CHR but neutrophils greater than or equal to 1.0 x 109/L and platelets greater than or equal to 20 x 109/L
without transfusions or bleeding.
Adult Patients With Chronic Phase
The MCyR rate in 321 CML-CP patients was 51%. The median time to MCyR among responders was 2.8 months (range,
1 to 28 months). The median duration of MCyR cannot be estimated. The median duration of exposure on this single
arm-trial was 18.4 months. Among the CML-CP patients who achieved MCyR, 62% of them had MCyR lasting more
than 18 months. The CCyR rate was 37%.
Adult Patients With Accelerated Phase
The overall confirmed hematologic response rate in 137 patients with CML-AP was 39%. The median time to first
hematologic response among responders was 1 month (range, 1 to 14 months). Among the CML-AP patients who
achieved HR, 44% of them had a response lasting for more than 18 months.
After imatinib failure, 24 different BCR-ABL mutations were noted in 42% of chronic phase and 54% of accelerated
phase CML patients who were evaluated for mutations.
14.3 Treatment Discontinuation in Newly Diagnosed Ph+ CML-CP Patients Who Have Achieved a Sustained
Molecular Response (MR4.5)
The ENEST freedom (Evaluating Nilotinib Efficacy and Safety in clinical Trials-freedom) study (NCT01784068) is an open-label, multicenter, single-arm study, where 215 adult patients with Ph+ CML-CP treated with nilotinib in first-line for ≥ 2 years who achieved MR4.5 as measured with the MolecularMD MRDx® BCR-ABL Test were enrolled to continue nilotinib treatment for an additional 52 weeks (nilotinib consolidation phase). Of the 215 patients, 190 patients (88.4%) entered the “Treatment-Free Remission” (TFR) phase after achieving a sustained molecular response (MR4.5) during the consolidation phase, defined by the following criteria:
• The 4 last quarterly assessments (taken every 12 weeks) were at least MR4 (BCR-ABL/ABL ≤ 0.01% IS), and
maintained for 1 year
• The last assessment being MR4.5 (BCR-ABL/ABL ≤ 0.0032% IS)
• No more than two assessments falling between MR4 and MR4.5 (0.0032% IS < BCR-ABL/ABL ≤ 0.01% IS).
The median age of patients who entered the TFR phase was 55 years, 49.5% were females, and 21.1% of the patients
were ≥ 65 years of age. BCR-ABL levels were monitored every 4 weeks during the first 48 weeks of the TFR phase.
Monitoring frequency was intensified to every 2 weeks upon the loss of MR4.0. Biweekly monitoring ended at one of
the following time points:
• Loss of MMR requiring patient to reinitiate nilotinib treatment
• When the BCR-ABL levels returned to a range between MR4.0 and MR4.5
• When the BCR-ABL levels remained lower than MMR for 4 consecutive measurements (8 weeks from initial loss of MR4.0).
Any patient with loss of MMR during the TFR phase reinitiated nilotinib treatment at 300 mg twice daily or at a reduced
dose level of 400 mg once daily if required from the perspective of tolerance, within 5 weeks after the collection date of
the blood sample demonstrating loss of MMR. Patients who required reinitiation of nilotinib treatment were monitored
for BCR-ABL levels every 4 weeks for the first 24 weeks and then every 12 weeks thereafter in patients who regained MMR.
Efficacy was based on the 96-week analysis data cut-off date, by which time, 91 patients (47.9%) discontinued from the
TFR phase due to loss of MMR, and 1 (0.5%), 1 (0.5%), 1 (0.5%) and 3 patients (1.6%) due to death from unknown
cause, physician decision, lost to follow-up and subject decision, respectively. Among the 91 patients who discontinued
the TFR phase due to loss of MMR, 88 patients restarted nilotinib treatment and 3 patients permanently discontinued
from the study.
By the 96-week data cut-off, of the 88 patients who restarted treatment due to loss of MMR in the TFR phase, 87 patients
(98.9%) patients regained MMR (one patient discontinued study permanently due to subject decision after 7.1 weeks of
retreatment without regaining MMR) and 81 patients (92.0%) regained MR4.5 by the time of the cut-off date. The
cumulative rate of MMR and MR4.5 regained at 24 weeks since treatment reinitiation was 97.7% (86/88 patients) and
86.4% (76/88 patients), respectively.
Table 14: Efficacy Results for ENEST freedom
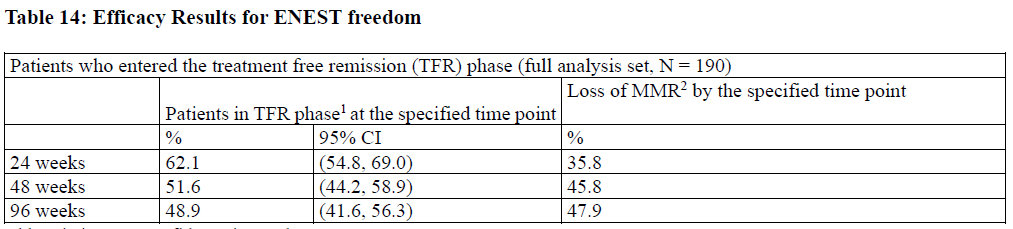
Abbreviation: CI, confidence interval. 1Patients in MMR at the specified time point in the TFR phase. 2Based on the time to event (loss of MMR) data during the TFR phase. Among the 190 patients in the TFR phase, 98 patients had a treatment-free survival (TFS) event (defined as discontinuation from TFR phase due to any reason, loss of MMR, death due to any cause, progression to AP/BC up to the end of TFR phase, or reinitiation of treatment due to any cause in the study) by the 96-week cut-off date.
Figure 1: Kaplan-Meier Estimate of Treatment-Free Survival After Start of TFR (Full Analysis Set ENEST freedom)
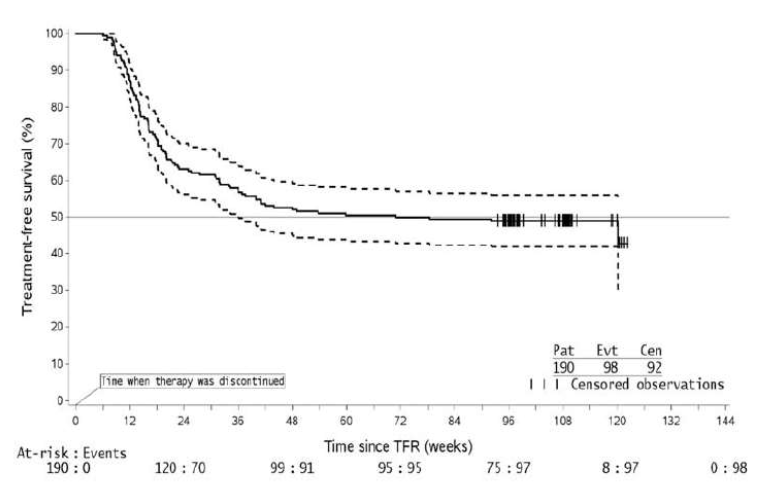
1. For a given time point, the points on the dashed curves represent the 95% confidence limits for the associated KM estimate on
the solid curve.
2. By the time of the 96-week data cut-off date, one single patient lost MMR at Week 120, at the time when only 8 patients were
considered at risk. This explains the artificial drop at the end of the curve.
14.4 Treatment Discontinuation in Ph+ CML-CP Patients Who Have Achieved a Sustained Molecular Response (MR4.5) on Nilotinib Capsules Following Prior Imatinib Therapy
The ENESTop (Evaluating Nilotinib Efficacy and Safety in clinical Trials-STop) study (NCT01698905) is an openlabel, multicenter, single-arm study, where 163 adult patients with Ph+ CML-CP taking tyrosine kinase inhibitors (TKIs) for ≥ 3 years (imatinib as initial TKI therapy for more than 4 weeks without documented MR4.5 on imatinib at the time of switch to nilotinib, then switched to nilotinib for at least 2 years), and who achieved MR4.5 on nilotinib treatment as measured with the MolecularMD MRDx® BCR-ABL Test were enrolled to continue nilotinib treatment for an additional 52 weeks (nilotinib consolidation phase). Of the 163 patients, 126 patients (77.3%) entered the TFR phase after achieving a sustained molecular response (MR4.5) during the consolidation phase, defined by the following criterion: • The 4 last quarterly assessments (taken every 12 weeks) showed no confirmed loss of MR4.5 (BCRABL/ABL ≤ 0.0032% IS) during 1 year. The median age of patients who entered the TFR phase was 56 years, 55.6% were females, and 27.8% of the patients were ≥ 65 years of age. The median actual dose intensity during the 52-week nilotinib consolidation phase was 771.8 mg/day with 52.4%, 29.4%, 0.8%, 16.7%, and 0.8% of patients receiving a daily nilotinib dose of 800 mg, 600 mg, 450 mg, 400 mg and 300 mg just before entry into the TFR phase, respectively. Patients who entered the TFR phase but experienced two consecutive measurements of BCR-ABL/ABL > 0.01% IS were considered having a confirmed loss of MR4.0, triggering reinitiation of nilotinib treatment. Patients with loss of MMR in the TFR phase immediately restarted nilotinib treatment without confirmation. All patients who restarted nilotinib therapy had BCR-ABL transcript levels monitored every 4 weeks for the first 24 weeks, then once every 12 weeks. Efficacy was based on the 96-week analysis data cut-off date, by which time, 61 patients (48.4%) had discontinued from the TFR phase: 58 patients (46.0%) due to loss of MMR or confirmed loss of MR4.0, 2 patients (1.6%) due to subject/guardian decision and one patient (0.8%) due to pregnancy. Among the 58 patients who discontinued from the TFR phase due to confirmed loss of MR4.0 or loss of MMR, 56 patients restarted nilotinib therapy and 2 patients permanently discontinued from the study. By the 96-week data cut-off, of the 56 patients who restarted nilotinib treatment due to confirmed loss of MR4.0 or loss of MMR in the TFR phase, 52 patients (92.9%) regained MR4.0 and MR4.5; 4 patients (7.1%) did not regain MR4.0 by the time of the cut-off date. The cumulative rate of MR4 and MR4.5 regained by 48-weeks since treatment reinitiation, was 92.9% (52/56 patients) and 91.1% (51/56 patients), respectively.
Table 15: Efficacy Results for ENESTop
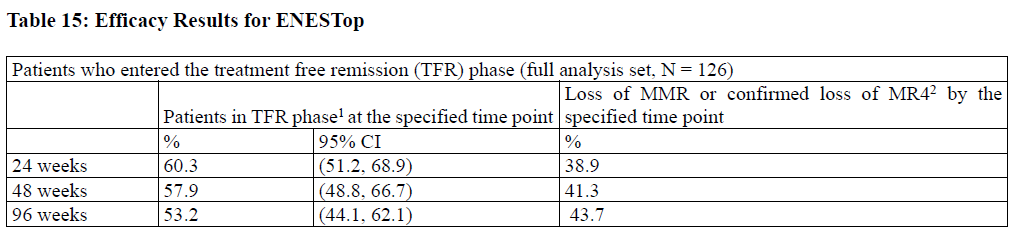
Abbreviation: CI, confidence interval. 1Patients without loss of MMR or confirmed loss of MR4 by specified time point of TFR phase. 2Based on the time to event (loss of MMR or confirmed loss of MR4) data during the TFR phase. Among the 126 patients in the TFR phase, 61 patients (48.4%) had a treatment-free survival (TFS) event (defined as discontinuation from TFR phase due to any reason, loss of MMR, confirmed loss of MR4, death due to any cause, progression to AP/BC up to the end of TFR phase, or reinitiation of treatment due to any cause in the study) on or before the 96-month cut-off date.
Figure 2: Kaplan-Meier Estimate of Treatment-Free Survival after Start of TFR (Full Analysis Set ENESTop)
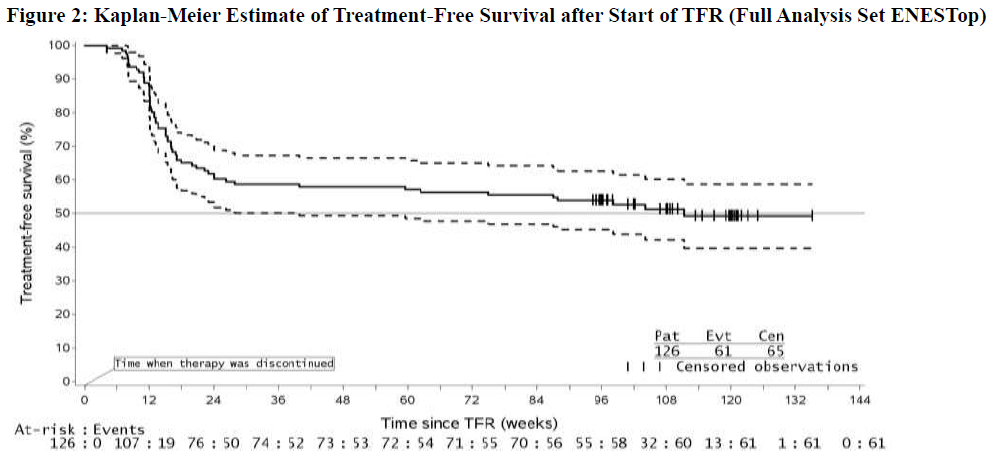
1. For a given time point, the points on the dashed curves represent the 95% confidence limits for the associated KM estimate on the solid curve. Additional pediatric use information is approved for Novartis Pharmaceuticals Corporation’s Tasigna® (nilotinib) capsules. However, due to Novartis Pharmaceuticals Corporation’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.

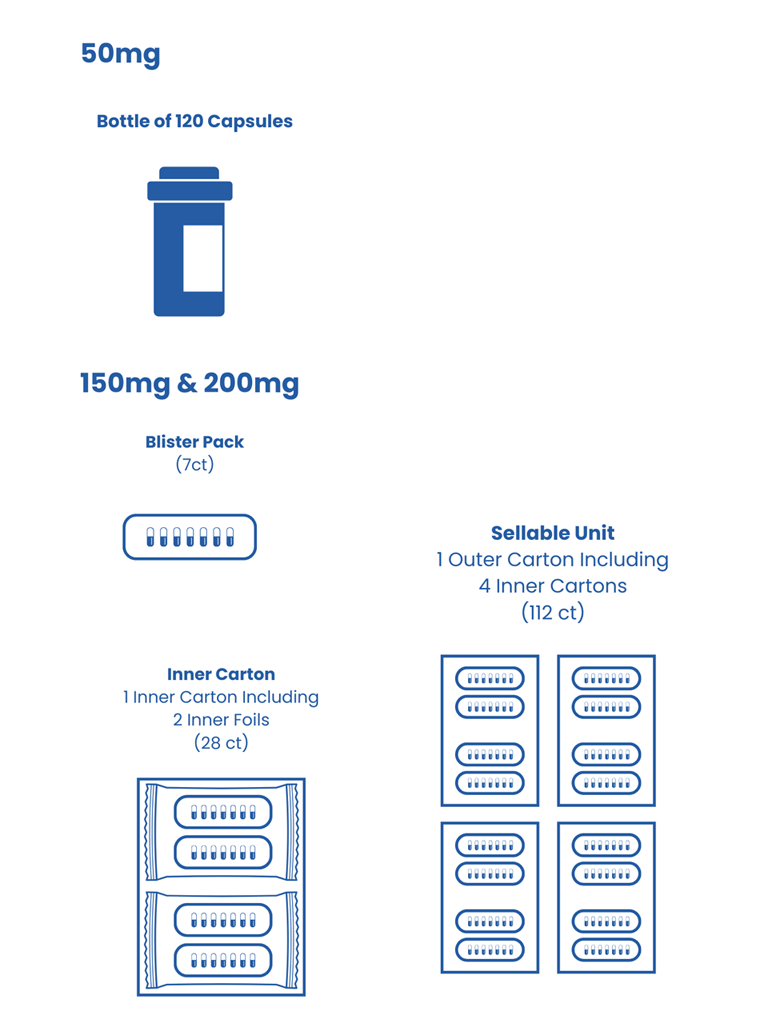
Nilotinib Capsule, 50 mg
Nilotinib Capsule, 150 mg

Nilotinib Capsule, 200 mg
Important Safety Information and Indications for Nilotinib Capsules
INDICATIONS
Adult patients with newly diagnosed Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase.
Adult patients with chronic phase (CP) and accelerated phase (AP) Ph+ CML resistant to or intolerant to prior therapy that included imatinib.
BOXED WARNING: QT PROLONGATION and SUDDEN DEATHS
- NILOTINIB prolongs the QT interval. Prior to NILOTINIB administration and periodically, monitor for hypokalemia or hypomagnesemia and correct deficiencies. Obtain ECGs to monitor
- the QTc at baseline, 7 days after initiation, and periodically thereafter, and following any dose adjustments
- Sudden deaths have been reported in patients receiving NILOTINIB. Do not administer NILOTINIB to patients with hypokalemia, hypomagnesemia, or long QT syndrome
- Avoid use of concomitant drugs known to prolong the QT interval and strong CYP3A4 inhibitors
- Avoid food 2 hours before and 1 hour after taking the dose
CONTRAINDICATIONS
Do not use in patients with hypokalemia, hypomagnesemia, or long QT syndrome.
WARNINGS AND PRECAUTIONS
Myelosuppression
Treatment with NILOTINIB can cause Grade 3/4 thrombocytopenia, neutropenia, and anemia. Perform complete blood counts every 2 weeks for the first 2 months and then monthly thereafter, or as clinically indicated. Myelosuppression was generally reversible and usually managed by withholding NILOTINIB temporarily or dose reduction.
QT Prolongation
NILOTINIB prolongs the QT interval. ECGs should be performed at baseline, 7 days after initiation, periodically as clinically indicated, and following dose adjustments. Correct hypokalemia or hypomagnesemia prior to administration and monitor periodically. Significant prolongation of the QT interval may occur when NILOTINIB is inappropriately taken with food and/or strong CYP3A4 inhibitors and/or medicinal products with a known potential to prolong QT. Therefore, co-administration with food must be avoided and concomitant use with strong CYP3A4 inhibitors and/or medicinal products with a known potential to prolong QT should be avoided. The presence of hypokalemia and hypomagnesemia may further enhance this effect.
Sudden Deaths
Sudden deaths have been reported in patients with Ph+ CML treated with NILOTINIB. The relatively early occurrence of some of these deaths relative to the initiation of NILOTINIB suggests the possibility that ventricular repolarization abnormalities may have contributed to their occurrence.
Cardiac and Arterial Vascular Occlusive Events
Cardiovascular events, including arterial vascular occlusive events, were reported in a randomized, clinical trial in patients with newly diagnosed Ph+ CML and observed in the postmarketing reports of patients receiving NILOTINIB therapy. Cases of cardiovascular events included ischemic heart disease-related events, peripheral arterial occlusive disease, and ischemic cerebrovascular events. If acute signs or symptoms of cardiovascular events occur, advise patients to seek immediate medical attention. The cardiovascular status of patients should be evaluated and cardiovascular risk factors should be monitored and actively managed during NILOTINIB therapy according to standard guidelines.
Pancreatitis and Elevated Serum Lipase
NILOTINIB can cause increases in serum lipase. Patients with a previous history of pancreatitis may be at greater risk of elevated serum lipase. If lipase elevations are accompanied by abdominal symptoms, interrupt dosing and consider appropriate diagnostics to exclude pancreatitis. Test serum lipase levels monthly or as clinically indicated.
Hepatotoxicity
NILOTINIB may result in hepatotoxicity as measured by elevations in bilirubin, AST/ALT, and alkaline phosphatase. Grade 3-4 elevations of bilirubin, AST, and ALT were reported at a higher frequency in pediatric patients than in adults. Monitor hepatic function tests monthly or as clinically indicated.
Electrolyte Abnormalities
The use of NILOTINIB can cause hypophosphatemia, hypokalemia, hyperkalemia, hypocalcemia, and hyponatremia. Correct electrolyte abnormalities prior to initiating NILOTINIB and monitor these electrolytes periodically during therapy.
Tumor Lysis Syndrome
Tumor lysis syndrome cases have been reported in patients taking NILOTINIB who have resistant or intolerant Ph+ CML. Malignant disease progression, high white blood cell counts, and/or dehydration were present in most of these cases. Maintain adequate hydration and correct uric acid levels prior to initiating therapy with NILOTINIB.
Hemorrhage
Serious hemorrhage, including fatal events, from any site, including the GI tract, was reported in patients with Ph+ CML receiving NILOTINIB. Monitor patients for signs and symptoms of bleeding and medically manage as needed.
Total Gastrectomy
Since the exposure of NILOTINIB is reduced in patients with total gastrectomy, perform more frequent monitoring of these patients. Consider dose increase or alternative therapy in patients with total gastrectomy.
Lactose
Since the capsules contain lactose, NILOTINIB is not recommended for patients with rare hereditary problems of galactose intolerance, severe lactase deficiency with a severe degree of intolerance to lactose-containing products, or of glucose-galactose malabsorption.
Monitoring Laboratory Tests
Complete blood counts should be performed every 2 weeks for the first 2 months and then monthly thereafter. Perform chemistry panels, including electrolytes, calcium, magnesium, liver enzymes, lipid profile, and glucose prior to therapy and periodically. ECGs should be obtained at baseline, 7 days after initiation, and periodically thereafter, as well as following dose adjustments.
Monitor lipid profiles and glucose periodically during the first year of NILOTINIB therapy and at least yearly during chronic therapy. Assess glucose levels before initiating treatment with NILOTINIB and monitor during treatment as clinically indicated. If test results warrant therapy, physicians should follow their local standards of practice and treatment guidelines.
Fluid Retention
Severe (Grade 3 or 4) fluid retention including pleural effusion, pericardial effusion, ascites, and pulmonary edema have been reported in patients with Ph+ CML receiving NILOTINIB. Monitor patients for signs of severe fluid retention (eg, unexpected rapid weight gain or swelling) and for symptoms of respiratory or cardiac compromise (eg, shortness of breath); evaluate etiology and treat patients accordingly.
Effects on Growth and Development in Pediatric Patients
Growth retardation has been reported in pediatric patients with Ph+ CML in chronic phase treated with NILOTINIB. Monitor growth and development in pediatric patients receiving NILOTINIB treatment.
Embryo-Fetal Toxicity
NILOTINIB can cause fetal harm. Advise females to inform their doctor if they are pregnant or become pregnant. Inform female patients of the risk to the fetus and potential for loss of the pregnancy. Advise females of reproductive potential to use effective contraception during treatment and for 14 days after receiving the last dose of NILOTINIB. Advise lactating women not to breastfeed during treatment with NILOTINIB and for at least 14 days after the last dose.
Monitoring of BCR-ABL Transcript Levels
Monitor BCR-ABL transcript levels in patients eligible for treatment discontinuation using an FDA-authorized test validated to measure molecular response (MR) levels with a sensitivity of at least MR4.5. In patients who discontinue NILOTINIB therapy, assess BCR-ABL transcript levels monthly for 1 year, then every 6 weeks for the second year, and every 12 weeks thereafter during treatment discontinuation. Following a loss of MMR (first line/second line) or confirmed loss of MR4 (2 consecutive measures separated by at least 4 weeks showing loss of MR4 in second line), patients should reinitiate NILOTINIB within 4 weeks of when the loss of remission is known to have occurred. Monitor CBC and BCR-ABL transcripts in patients who reinitiate treatment with NILOTINIB due to loss of MR quantitation every 4 weeks until MMR is reestablished and then every 12 weeks. For patients who fail to achieve MMR after 3 months of treatment reinitiating, BCR-ABL kinase domain mutation testing should be performed.
ADVERSE REACTIONS
The most commonly reported nonhematologic adverse reactions (≥20%) in adult and pediatric patients receiving NILOTINIB were nausea, rash, headache, fatigue, pruritus, vomiting, diarrhea, cough, constipation, arthralgia, nasopharyngitis, pyrexia, and night sweats. Hematologic adverse drug reactions (all grades) include myelosuppression: thrombocytopenia, neutropenia, and anemia. Musculoskeletal symptoms (eg, myalgia, pain in extremity, arthralgia, bone pain, spinal pain, or musculoskeletal pain) have been reported in eligible patients who discontinued NILOTINIB therapy after attaining a sustained MR4.5. The rate of new musculoskeletal symptoms (all grades) generally decreased from the first year (34%-48%) to the second year (9%-15%) after treatment discontinuation.
DOSE ADJUSTMENTS OR MODIFICATIONS
NILOTINIB may need to be temporarily withheld and/or dose reduced for QT prolongation, hepatic impairment, hematologic toxicities that are not related to underlying leukemia, clinically significant moderate or severe nonhematologic toxicities, laboratory abnormalities (lipase, amylase, bilirubin, or hepatic transaminase elevations) or concomitant use of strong CYP3A4 inhibitors.
DRUG INTERACTIONS
Avoid concomitant use of strong CYP3A4 inhibitors with NILOTINIB, or reduce NILOTINIB dose if co-
administration cannot be avoided. Avoid concomitant use of strong CYP3A4 inducers with NILOTINIB. Use
short-acting antacids or H2 blockers as an alternative to proton pump inhibitors. Avoid coadministration of
NILOTINIB with agents that may prolong the QT interval, such as anti-arrhythmic drugs.
Please see full Prescribing Information, including Boxed WARNING, by clicking here.
To report SUSPECTED ADVERSE REACTIONS, contact Cipla Ltd. at 1-866-604-3268 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DIRECTIONALS TO THE PI:
Please see Important Safety Information and the full Prescribing Information, including Boxed Warning.